
Strukturer
Reaktionskinetik
Hvad går det ud på?
Når vi taler om strukturerer ifm. reaktionskinetikken, er der reelt to typer struktur der er vigtig: den fysiske form og molekylstrukturen.
I de simple udgaver af reaktionskinetikken, ser vi kun på de kemiske forbindelser som kugler der støder sammen og reagerer, f.eks. fældningsreaktionen
Ag+(aq) + Cl−(aq) AgCl(s)
AgCl(s)
som er yderst populær i undervisningssammenhæng. Ionerne Ag+ og Cl− er sfæriske, så der er ikke noget forkert ved modellen, den er blot ikke så repræsentativ som man kunne få indtryk af.
I den virkelige verden, og især når vi bevæger os væk fra den klassiske, uorganiske kemi, begynder molekylernes arkitektur hurtigt at spille en stor rolle, og det samme gør tilstandsformer og partikelmorfologier.
Hvis vi ser på reaktionen:
A + B-C A-B +C
så må molekylet A skulle ramme B på B-C-molekylet for at kunne binde sig til dette. Dette findes i flere versioner hvor A rammer B så C skilles fra eller hvor C skilles fra B først, så B kan reagere med A. De to reaktionstyper SN1 og SN2 er netop disse to scenarier.
Alene det at A og B skal ramme hinanden, påvirker reaktionshastigheden fordi det er et spørgsmål om sandsynligheder. Ud over at skulle ramme hinanden, skal de således også ramme fra den rigtige vinkel, endnu et spørgsmål om sandsynligheder, og ud over at skulle ramme fra den rigtige vinkel, skal A og B også ramme sammen med tilstrækkelig kraft. Vi kan derfor skrive reaktionshastigheden som
Tilstrækkelig energi ved kollisionerne er det der hedder aktiveringsenerien Ea, som bliver gennemgået i et separat kapitel.
I de simple udgaver af reaktionskinetikken, ser vi kun på de kemiske forbindelser som kugler der støder sammen og reagerer, f.eks. fældningsreaktionen
Ag+(aq) + Cl−(aq)
AgCl(s)som er yderst populær i undervisningssammenhæng. Ionerne Ag+ og Cl− er sfæriske, så der er ikke noget forkert ved modellen, den er blot ikke så repræsentativ som man kunne få indtryk af.
I den virkelige verden, og især når vi bevæger os væk fra den klassiske, uorganiske kemi, begynder molekylernes arkitektur hurtigt at spille en stor rolle, og det samme gør tilstandsformer og partikelmorfologier.
Hvis vi ser på reaktionen:
A + B-C
A-B +Cså må molekylet A skulle ramme B på B-C-molekylet for at kunne binde sig til dette. Dette findes i flere versioner hvor A rammer B så C skilles fra eller hvor C skilles fra B først, så B kan reagere med A. De to reaktionstyper SN1 og SN2 er netop disse to scenarier.
Alene det at A og B skal ramme hinanden, påvirker reaktionshastigheden fordi det er et spørgsmål om sandsynligheder. Ud over at skulle ramme hinanden, skal de således også ramme fra den rigtige vinkel, endnu et spørgsmål om sandsynligheder, og ud over at skulle ramme fra den rigtige vinkel, skal A og B også ramme sammen med tilstrækkelig kraft. Vi kan derfor skrive reaktionshastigheden som
| v = | Antal kollisioner mellem A og B-C pr. tidsenhed | × | Fraktionen af kollisioner med korrekt orientering | × | Fraktionen af kollisioner med tilstrækkelig energi til at kunne give en reaktion |
Tilstrækkelig energi ved kollisionerne er det der hedder aktiveringsenerien Ea, som bliver gennemgået i et separat kapitel.
Antal kollisioner
Antal kollisioner afhænger af flere faktorer, hvoraf en del er de fysiske strukturer og tilstandsformer, eller hænger sammen med disse.
- Koncentrationerne af de kemiske forbindelser der skal reagere. Højere koncentrationer øger sandsynligheden for kollisioner. I gasfase har man en mulighed for at øge koncentrationen yderligere, ved at hæve trykket og på den måde tvinge de kemiske forbindelser tættere på hinanden. Øger man trykket, skal man være opmærksom på, at gasserne ikke længere kan forventes at opføre sig som idealgasser, dvs. der kan opstå uventede eller dominerende sidereaktioner der påvirker udbyttet i negativ retning.
- Bevægeligheden af de kemiske forbindelser. Vigtigheden af dette stiger med faldende koncentrationer, fordi molekylerne rent statistisk skal bevæge sig længere for at møde modparten de skal reagere med. Her bliver temperatur en vigtig faktor, fordi de kemiske forbindelser i gas- og væskefase bevæger sig hurtigere og dermed længere pr. tidsenhed, ved øgede temperaturer. Procesteknisk hjælper man dette på vej ved at skabe turbulens i blandingerne.
- Tilgængelighed, når vi arbejder med flerfasesystemer. Hvis der er tale om faseseparerede reaktanter, vil reaktionen ske på kontaktfladerne. I den praktiske kemi ser vi det ofte kun nævnt som øget reaktivitet ved mindre partikelstørrelse på faste stoffer, dvs. større overflade, men dette er en forsimpling. Partikelmorfologien spiller også ind. En kugle har det mindste overfladeareal pr. volumenenhed, hvor et pladeformet eller nåleformet materiale vil have en langt større overflade pr. volumenenhed. Partikler i denne sammenhæng er ikke kun faste stoffer, det indbefatter også sådan noget som aerosoler og dispersioner, hvor der er tale om væskepartikler i en gasfase eller en anden væskefase. Under ideale forhold er gasser miscible, men under forhold hvor gasserne ikke opfører sig som idealgasser, f.eks. under tryk hvor de kemiske forbindelsers elektrostatiske kræfter begynder at spille ind, vil man også se effekter svarende til faseseparering.
Molekylernes orienteringen og struktur
I væske- og gasfase vil molekylerne i et eller andet omfang rotere om sin egen akse. Hvordan og hvor hurtigt afhænger af både molekylets struktur og dets omgivelser. Skal man have molekylet A til at ramme B på B-C-molekylet så de reagerer, skal det ramme fra den rigtige vinkel:
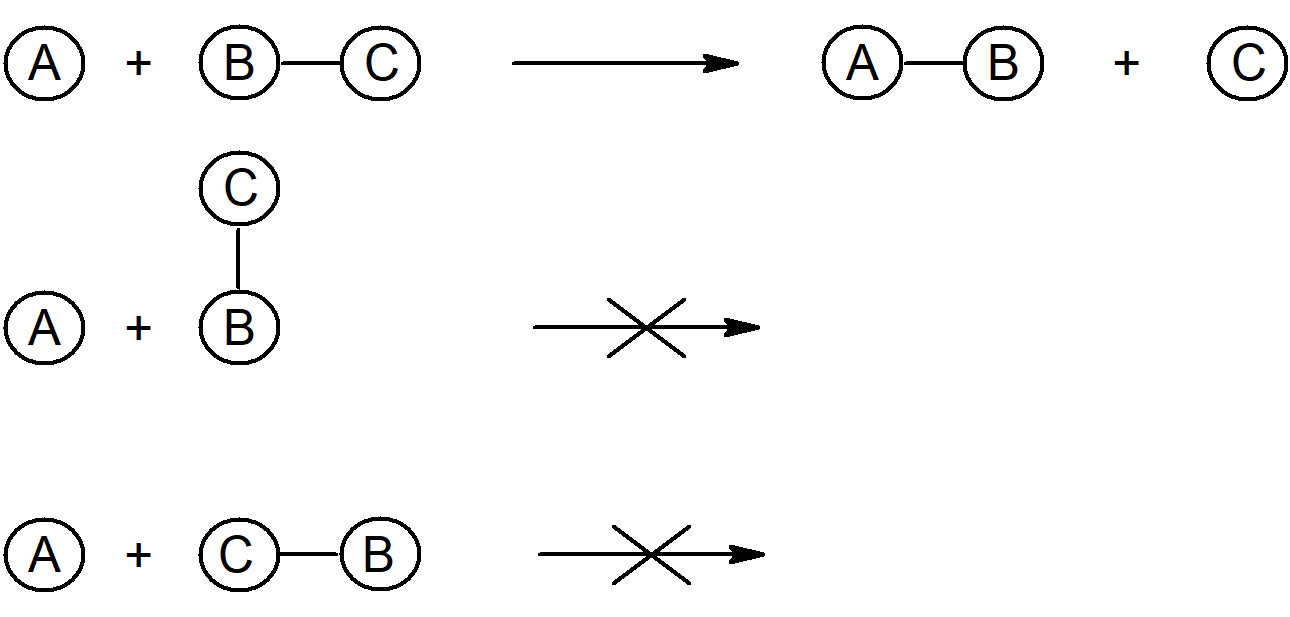
At ramme fra den rigtige vinkel, kræver at der er plads, hvilket bringer os til problemet med steriske hindringer. Atomerne fylder og sidder i vejen for reaktionerne. Den viste reaktionsmekanisme har yderligere en udfordring, fordi C skal afstødes. Det vender vi tilbage til.
Et godt eksempel på steriske hindringer er estersyntesen, som er en kondensationsreaktion. Syre plus alkohol, under sure omstændigheder, giver ester plus vand. For de primære alkoholer går det relativt let. For de sekundære alkoholer går det knapt så let, så reaktionshastigheden falder. For de tertiære alkoholer falder reaktionshastigheden til næsten nul. Når vi tegner det med vores almindelige håndtegninger, f.eks. disse tre variationer over en alkohol på fire carbon:
kan det være svært at se hvorfor det lige skulle være så svært at reagere en syre med en tertiær alkohol, men det er fordi denne type tegninger give et optisk bedrag af placeringer og størrelsesforhold. Hvis vi i stedet ser de tre alkoholer i 3D med space filling, så vi kan se alkoholmolekylernes korrekte arkitektur, bliver det mere synligt, hvor meget tilgængeligheden af alkoholgruppen reduceres ved en tertiær alkohol:
En udfordring der ligger i substitutionsreaktioner, og dette er både de nucleofile og de elektrofile, er, at når molekylet A skal ramme B på B-C-molekylet, skal C kunne gå af. Den gruppe der går af hedder en leaving group. Leaving groups skal derfor ikke sidde for godt fast, for ellers har de svært ved at gå fra, når substituenten skal sættes på. Halogener (fluor, chlor, brom og iod) er gode leaving groups, alkoholer (-OH) er relativt dårlige, aminer (-NH2) er endnu værre. Alt sammen noget strukturelt der påvirker reaktionshastigheden.
En af de udfordringer man også kan stå med, med de kemiske reaktioner er, at molekylere kan have flere konformationer. Det eksempel ser ofte trækkes frem er cyclohexan, hvor man har stolformen og bådformen:
Man taler derfor om det der hedder den sterisk favorable konformation og den energetisk favorable konformation, og de behøver ikke at være det samme. Der kan for nogle molekyler, især ved makromolekylerne, godt være flere energetisk favorable konformationer. Hvis det kun er den ene konformation der er brugbar ved reaktionerne, vil man opleve dette som en temperatur-intervaller man er begrænset af.
En måde at påvirke konformationen, som ikke er temperatur, er solventer og katalysatorer. Når det kommer til solventer, kan man arbejde med både polaritet, pH og ionstyrke, afhængig af reaktionen. Et kendt eksempel er DNA, der under normale forhold findes i den helix-formation der hedder B-DNA, som er stabiliseret af hydrogenbindinger og vandmolekyler i det der hedder major groove. Hvis man reducerer vandmængden, skifter DNA konformation til en mere kompakt form, der hedder A-DNA, og hæver man ionstyrken i vandet, kan man få DNA-dobbelthelixen til at dreje den modsatte vej rundt i den konformation, der hedder Z-DNA.
Ofte er det man arbejder med, når man arbejder med solventer og katalysatorer, en stabilisering af transition state eller en favorabel konformation, men det kan også, for katalysatorernes vedkommende, være en måde at fiksere orienteringen eller placeringen af reaktanterne. Den kommercielle metode til hydrogenering af dobbeltbindinger ved at have H2 i en platin- eller palladiumkatalysator er netop, at man fikserer H2 på metaloverfladen og gør den lettere tilgængelig for de dobbeltbindinger der skal hydrogeneres.
At ramme fra den rigtige vinkel, kræver at der er plads, hvilket bringer os til problemet med steriske hindringer. Atomerne fylder og sidder i vejen for reaktionerne. Den viste reaktionsmekanisme har yderligere en udfordring, fordi C skal afstødes. Det vender vi tilbage til.
Et godt eksempel på steriske hindringer er estersyntesen, som er en kondensationsreaktion. Syre plus alkohol, under sure omstændigheder, giver ester plus vand. For de primære alkoholer går det relativt let. For de sekundære alkoholer går det knapt så let, så reaktionshastigheden falder. For de tertiære alkoholer falder reaktionshastigheden til næsten nul. Når vi tegner det med vores almindelige håndtegninger, f.eks. disse tre variationer over en alkohol på fire carbon:
| 1-buthanol | 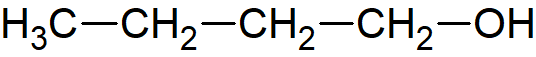 |
| 2-buthanol | 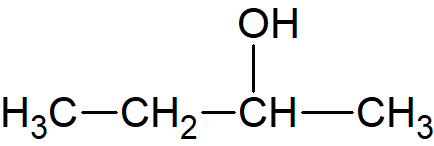 |
| tert-butanol | 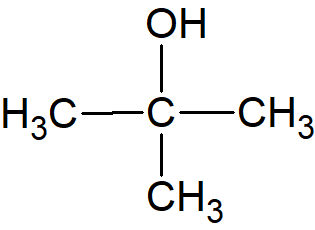 |
kan det være svært at se hvorfor det lige skulle være så svært at reagere en syre med en tertiær alkohol, men det er fordi denne type tegninger give et optisk bedrag af placeringer og størrelsesforhold. Hvis vi i stedet ser de tre alkoholer i 3D med space filling, så vi kan se alkoholmolekylernes korrekte arkitektur, bliver det mere synligt, hvor meget tilgængeligheden af alkoholgruppen reduceres ved en tertiær alkohol:
| 1-buthanol | 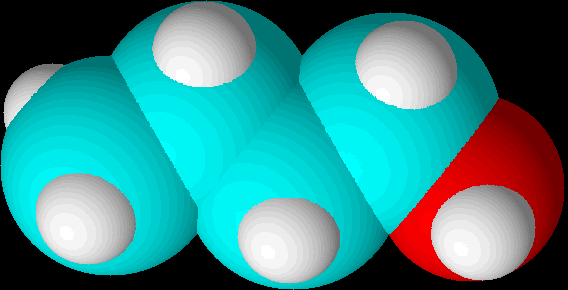 |
| 2-buthanol | 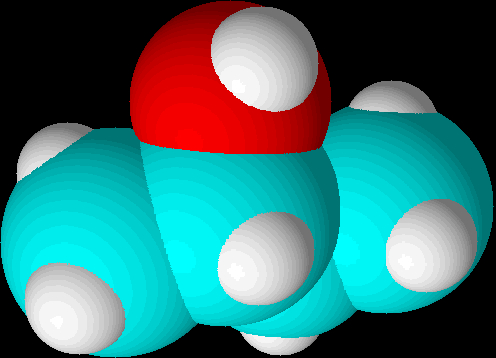 |
| tert-butanol | 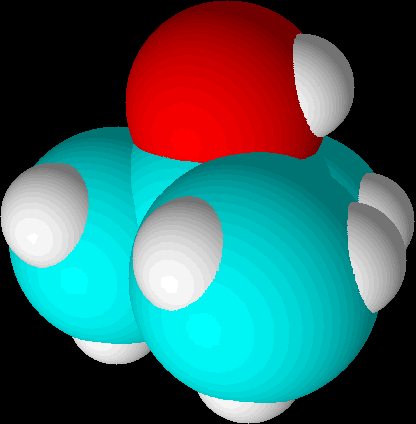 |
En udfordring der ligger i substitutionsreaktioner, og dette er både de nucleofile og de elektrofile, er, at når molekylet A skal ramme B på B-C-molekylet, skal C kunne gå af. Den gruppe der går af hedder en leaving group. Leaving groups skal derfor ikke sidde for godt fast, for ellers har de svært ved at gå fra, når substituenten skal sættes på. Halogener (fluor, chlor, brom og iod) er gode leaving groups, alkoholer (-OH) er relativt dårlige, aminer (-NH2) er endnu værre. Alt sammen noget strukturelt der påvirker reaktionshastigheden.
En af de udfordringer man også kan stå med, med de kemiske reaktioner er, at molekylere kan have flere konformationer. Det eksempel ser ofte trækkes frem er cyclohexan, hvor man har stolformen og bådformen:
| cyclohexan stolform |  |
| cyclohexan bådform | 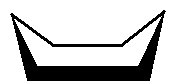 |
Man taler derfor om det der hedder den sterisk favorable konformation og den energetisk favorable konformation, og de behøver ikke at være det samme. Der kan for nogle molekyler, især ved makromolekylerne, godt være flere energetisk favorable konformationer. Hvis det kun er den ene konformation der er brugbar ved reaktionerne, vil man opleve dette som en temperatur-intervaller man er begrænset af.
En måde at påvirke konformationen, som ikke er temperatur, er solventer og katalysatorer. Når det kommer til solventer, kan man arbejde med både polaritet, pH og ionstyrke, afhængig af reaktionen. Et kendt eksempel er DNA, der under normale forhold findes i den helix-formation der hedder B-DNA, som er stabiliseret af hydrogenbindinger og vandmolekyler i det der hedder major groove. Hvis man reducerer vandmængden, skifter DNA konformation til en mere kompakt form, der hedder A-DNA, og hæver man ionstyrken i vandet, kan man få DNA-dobbelthelixen til at dreje den modsatte vej rundt i den konformation, der hedder Z-DNA.
Ofte er det man arbejder med, når man arbejder med solventer og katalysatorer, en stabilisering af transition state eller en favorabel konformation, men det kan også, for katalysatorernes vedkommende, være en måde at fiksere orienteringen eller placeringen af reaktanterne. Den kommercielle metode til hydrogenering af dobbeltbindinger ved at have H2 i en platin- eller palladiumkatalysator er netop, at man fikserer H2 på metaloverfladen og gør den lettere tilgængelig for de dobbeltbindinger der skal hydrogeneres.